(a cura di Roberto Bigoni)
La termodinamica teorica deduce molti dei suoi risultati più significativi a livello macroscopico e microscopico dall'analisi del comportamento asintotico dei gas in condizioni di alta temperatura e bassa densità. Questo comportamento asintotico risulta uguale per tutte le sostanze e rende quindi possibile la definizione di leggi non specifiche per le singole sostanze ma valide in generale. Il comportamento asintotico è rappresentato dal modello del gas perfetto (o ideale), in cui, per un gas omogeneo (cioè non una miscela di vari gas, come, ad esempio, l'aria) si assume che:
Le proprietà fondamentali del gas perfetto sono espresse dall'equazione di stato dei gas perfetti
![]()
in cui P e V rappresentano pressione del gas e volume del recipiente, n il numero di moli, R la costante dei gas perfetti e T la temperatura assoluta.
Tuttavia le condizioni di temperatura e densità dei gas reali, in natura e nelle applicazioni tecnologiche, si discostano spesso dalle condizioni asintotiche e e questa relazione non ne rispecchia adeguatamente il comportamento.
Un adattamento dell'equazione di stato dei gas perfetti alla fenomenologia dei gas reali fu proposto dal fisico olandese J. D. van der Waals che propose di completarla in due aspetti.
Le particelle interagiscono elettricamente tra di loro, attirandosi l'una con l'altra con forze di origine elettrica dette forze di van der Waals. Dunque sono dotate, oltre che di energia cinetica, anche di energia potenziale, che, in quanto dovuta a forze attrattive, è negativa.
Una particella interna alla massa del gas è attratta in modo simmetrico dalle particelle circostanti: la forza risultante è nulla.
Ma una particella prossima alla superficie del recipiente è circondata da altre particelle solo nella parte interna alla superficie: la forza risultante non è nulla, è diretta perpendicolarmente alla superficie, verso l'interno.
Quindi le particelle che si avvicinano alla superficie sono frenate da quelle più interne, perdono velocità e quantità di moto e la pressione risulta minore di quella che si avrebbe, a parità di temperatura e di densità, in un gas perfetto.
La diminuzione di pressione è proporzionale sia alla pressione sia alla densità e dato che, a sua volta, la pressione è proporzionale alla densità, in definitiva tale diminuzione è proporzionale al quadrato della densità e conseguentemente al quadrato della densità molare.
Quindi per ottenere la pressione di un gas ideale nelle stesse condizioni, la pressione di un gas reale va aumentata di una quantità proporzionale al quadrato della densità molare.
Sulla base delle precedenti considerazioni l'equazione (1) va modificata nel seguente modo:
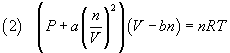
La (2) è detta equazione di van der Waals.
I parametri a e b di questa equazione sono specifici di ogni gas e vanno ricavati sperimentalmente.
Esempio.
Per il diossido di carbonio CO2 (anidride carbonica) si ha:
Si considerano 10 moli di CO2 contenute in 1 litro alla temperatura di 300K.
Se il CO2 si comportasse come un gas perfetto, dall'equazione (1) la sua pressione sarebbe
![]()
Applicando l'equazione (2) si ottiene invece
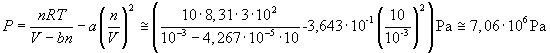
Si ottiene una pressione che è circa il 28% di quella precedente.
La (1) implica che le isoterme dei gas perfetti nel piano VP sono rappresentate da rami di iperbole equilatera. Se, ad esempio, si assume n=1 e si misurano il volume in litri e la pressione in atmosfere, per un gas perfetto le isoterme alle temperature di 300K, 400K, 500K e 600K sono
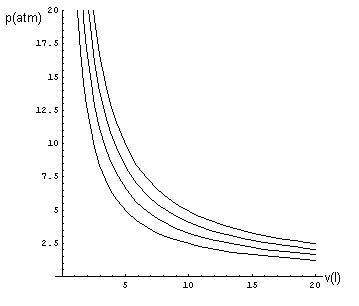
Se si graficano le isoterme dei gas reali deducendole dalla (2), per alte temperature non si notano sensibili differenze rispetto alle isoterme ideali, ma all'abbassarsi della temperatura, i grafici ottenuti si differenziano sempre più da quelle. Ad esempio, per 10 moli di CO2, alle temperature di 260K, 270K, 280K, 290K, 300K, 340K, si hanno le seguenti curve
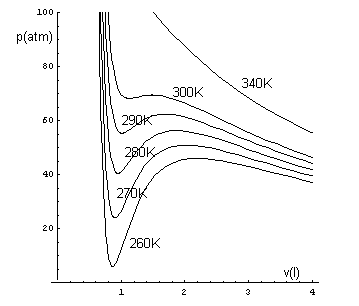
Su questi grafici si possono fare le seguenti osservazioni.
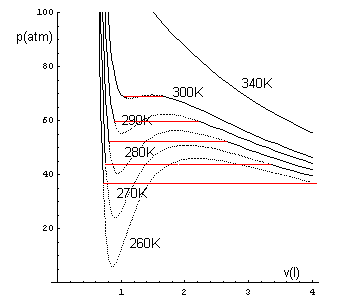
Quindi l'andamento effettivo delle isoterme è il seguente
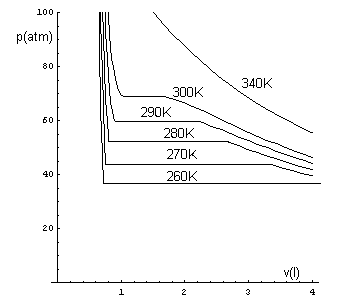
La zona di pressione costante in ogni isoterma rappresenta stati del sistema in cui la fase gassosa e la fase liquida sono compresenti. In questi stati c'è equilibrio dinamico tra le due fasi: in ogni intervallo di tempo, tante sono le particelle che passano dalla fase gassosa alla fase liquida quante quelle che fanno il passaggio inverso. La pressione di questi stati è detta pressione di vapor saturo (PVS).
Se si uniscono con una linea continua gli estremi dei tratti orizzontali delle isoterme (in rosso nella figura) e poi si traccia l'isoterma per il massimo di questa curva, detta isoterma critica che per il CO2 è circa 304K (in blu nella figura), il piano di Clapeyron viene diviso in quattro zone:
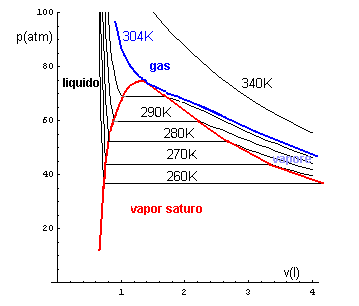
La temperatura dell'isoterma critica è detta temperatura critica TC. Al di sopra di questa temperatura il gas non può essere liquefatto.
Data la temperatura critica, la PVS corrispondente è detta pressione critica PC. Per il CO2 si ha PC ≅ 73 atm.
Per temperature inferiori alla TC la PVS diminuisce e il gas può essere liquefatto con pressioni inferiori alla PC.
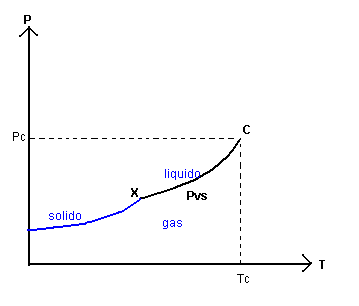
Data la TC e la PC, per temperature inferiori alla TC nel piano TP si può rappresentare l'andamento della PVS in funzione della temperatura assoluta T. Come s'è visto nel paragrafo precedente, al decrescere di T decresce anche la PVS, cioè la pressione di equilibrio tra fase gassosa e fase liquida. L'andamento della PVS in funzione di T è rappresentato qualitativamente dal seguente grafico
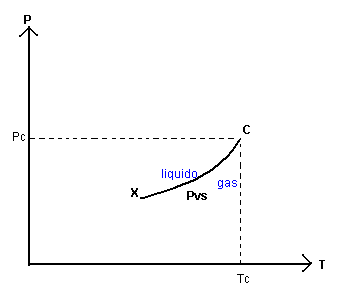
Il grafico della PVS si interrompe in un punto X perché per temperature inferiori all'ascissa di questo punto la sostanza non può esistere nella fase liquida: l'energia cinetica media delle particelle che la compongono diviene troppo bassa rispetto all'enegia potenziale e le posizioni delle particelle si fissano rigidamente: la sostanza passa alla fase solida. Al di sotto della temperatura di X esistono solo la fase solida ed eventualmente la fase gassosa per le particelle che hanno un energia cinetica molto maggiore di quella media.
Ovviamente dovranno esistere anche stati di frontiera tra la fase solida e la fase liquida e il diagramma delle fasi sarà completato nel seguente modo
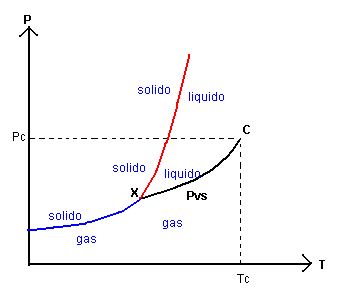
Lo stato X in cui possono coesistere tutte e tre le fasi di aggregazione della sostanza è detto punto triplo della sostanza.
Per il CO2 il punto triplo si ha a 216.5K e a 5.11atm.
Per l'acqua il punto triplo si ha a 273.16K e a 6.04·10-3atm.
Ognuna delle linee di frontiere dell'ultimo diagramma può essere attraversata nei due sensi. Per le corrispondenti transizioni di fase si assume la seguente nomenclatura.
| fase iniziale | fase finale | denominazione |
|---|---|---|
| gas | liquido | condensazione |
| liquido | gas | vaporizzazione |
| liquido | solido | cristallizzazione |
| solido | liquido | fusione |
| solido | gas | sublimazione |
| gas | solido | brinazione |
Se si riesamina una delle isoterme per 10 moli di CO2, ad esempio quella a 290K,
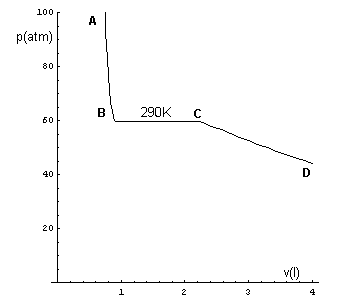
si osserva che nella dilatazione BC, che porta il sistema dalla fase liquida alla fase di vapore, il sistema compie un lavoro misurato, nel piano VP, dall'area sottesa dal segmento BC.
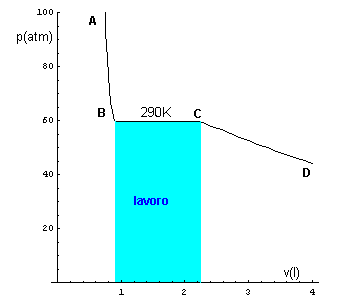
Come nelle isoterme dei gas ideali, questo lavoro senza diminuzione di temperatura è possibile solo se il sistema riceve calore dall'esterno, ma nelle sostanze reali, diversamente che nei gas ideali, il calore ricevuto deve compensare non solo il lavoro svolto ma anche il mantenimento della pressione. È quindi necessaria una dose supplementare di calore, detta calore latente di vaporizzazione che, a livello microscopico, può essere interpretata come l'energia necessaria per 'slegare' le particelle le une dalle altre, cioè perché la loro energia potenziale da negativa (particelle legate) diventi nulla (particelle libere).
Ad esempio, il calore latente di vaporizzazione del CO2 è 25.13 kJ/mole, quello dell'acqua è 40.8 kJ/mole.
Per avere una valutazione intuitiva della grandezza del calore latente di vaporizzazione, si noti che, dato che l'acqua ha un calore specifico di 4186 J/kg·K, per scaldare un litro di acqua da 0°C a 100°C occorrono 418600 J di calore. Per farla evaporare tutta, ricordando che una mole d'acqua sono circa 18g e che quindi un litro contiene circa 55.5 moli, occorrono ≅ 55.5·40800 J ≅ 2264000 J, cioè più del quintuplo del precedente.
La transizione di fase inversa CB (condensazione) che porta una sostanza dalla fase di vapore alla fase liquida, implica una diminuzione isoterma e isobara di volume. Il questa transizione vengono restituiti tutto il lavoro e tutto il calore assorbiti nella transizione BC e quello che prima appariva come calore latente di vaporizzazione ora viene emesso come calore latente di condensazione.
Le altre transizioni di fase sono isoterme e isobare come quella considerata ma anche, con buona approssimazione, isocore. Infatti tra liquidi e solidi le variazioni di volume sono di piccola entità. Il calore che compensa le variazioni di energia potenziale delle particelle è detto, a seconda dei casi, calore latente di fusione o cristallizzazione, di sublimazione o di brinazione.
La transizione di una sostanza da liquido a vapore non avviene solo nelle condizioni sopra descritte, cioè in presenza di vapor saturo. Se, come spesso succede, una porzione della superficie che delimita il liquido lo separa da un gas o dal vuoto (superficie libera), questa porzione di superficie è sede di un continuo scambio di particelle dalla fase liquida a quella di vapore e viceversa.
Le particelle della fase liquida, in quanto legate elettricamente alla massa delle particelle circostanti, hanno energia potenziale negativa. Ciò significa che sono in una 'buca di potenziale' dalla quale possono uscire solo se la loro energia cinetica supera il valore assoluto dell'energia potenziale. L'energia cinetica media è proporzionale alla temperatura assoluta ed è, nella fase liquida, minore del valore assoluto dell'energia potenziale. La maggioranza delle particelle non può così abbandonare la massa del liquido. Ma il fatto che l'energia cinetica media abbia un certo valor medio implica che una certa quota di particelle deve avere un'energia cinetica superiore alla media e tra queste alcune avranno energia cinetica superiore al valore assoluto dell'energia potenziale e, se sono prossime alla superficie libera, potranno uscire dalla buca, cioè passare alla fase aeriforme.
Questa modalità di vaporizzazione è detta evaporazione.
Se il volume che delimita le due fasi è molto grande e le particelle passate alla fase di vapore vengono allontanate dalla superficie libera si può avere la completa evaporazione del liquido.
Se il volume che delimita le due fasi è chiuso o comunque le particelle in fase di vapore non sono allontanate dalla superficie libera del liquido, alcune delle particelle di vapore che si trovino in prossimità della superficie libera potranno avere energia cinetica minore del valore assoluto dell'energia potenziale e ricadere nella buca, cioè tornare alla fase legata liquida.
Dopo un certo tempo questi due processi si equilibrano e le due fasi sono compresenti in proporzioni costanti. In questo stato di equilibrio il vapore è saturo e l'evaporazione non fornisce più contributi alla vaporizzazione.
Quando il vapore è saturo e si fornisce calore al sistema, la vaporizzazione avviene all'interno di tutta la massa liquida, cioè in tutto il volume occupato dal liquido ci sono particelle che passano allo stato di vapore, formando bollicine di gas che esercita pressione contro il liquido circostante. Ma se la pressione del liquido è maggiore della pressione di vapor saturo, queste bollicine sono schiacciate, implodono, e il gas in esse contenuto ritorna in fase liquida e l'energia delle particelle contenute in esse contribuisce a riscaldare il liquido.
Quando invece la pressione del vapor saturo nelle bollicine uguaglia la pressione del liquido circostante esse permangono, per il principio di Archimede salgono alla superficie e il gas in esse contenuto permane nello stato aeriforme. In questo caso si ha l'ebollizione del liquido.
Quindi la vaporizzazione per ebollizione può avvenire solo se la pressione del vapor saturo uguaglia o supera la pressione del liquido circostante. E dato che la pressione del liquido, per profondità non troppo elevate, coincide con la pressione sulla superficie libera dello stesso, si ha ebollizione solo quando la pressione di vapor saturo eguaglia la pressione sulla superficie libera.
Se la superficie libera è a contatto con l'atmosfera, si ha ebollizione solo quando la pressione di vapor saturo eguaglia la pressione atmosferica. Se la pressione esterna è esattamente di una atmosfera, la temperatura alla quale la pressione di vapor saturo dell'acqua è quella che è stata fissata in 100°C, ma se le pressione esterna è minore, la temperatura di ebollizione dell'acqua cala.
Viceversa, se si scalda acqua in un recipiente chiuso, il vapore che si forma esercita sulla superficie libera una pressione tanto maggiore, quanto maggiore è la sua temperatura (legge delle isocore) per cui occorrerà una temperatura maggiore perché le bollicine di vapore all'interno della massa liquida non implodano e si abbia quindi l'ebollizione. Questo è, ad esempio, il principio in base al quale funzionano le pentole a pressione.
ultima revisione: Maggio 2018